Project 1.4
Comprehensive characterization of microbiota associated with InsP6 degradation in the gastrointestinal tract of laying hens and quails
Amélia Camarinha-Silva / Jana Seifert
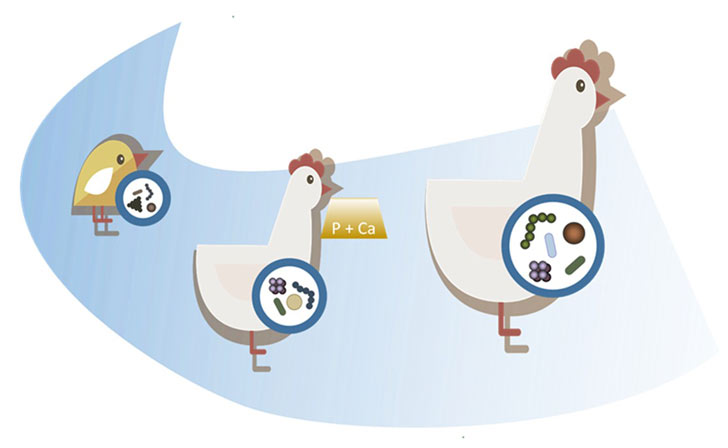
This project aims to contribute to the understanding of the composition and function of the intestinal microbiota in laying hens and quails. The focus is set on the impact of InsP6 degradation products and the productive life span as factors contributing to variations of the intestinal microbiota.
Furthermore, through sharing samples obtained in the due course of the project and also methods with other partners of the Research Unit, the project aims to improve the understanding of the fundamental principles of microbe-host interactions in single animals. The microbiota is studied using RNA extracts of intestinal samples and sequence analyses of the 16S rRNA gene as well as the metatranscriptome. Additionally, concomitant samples are used to analyze the metaproteome and metabolome to study the influence towards the microbiome functions.