Project 1.2
Analysis of P utilization using the host genome and microbiota variability in Japanese quail
Jörn Bennewitz / Amélia Camarinha-Silva

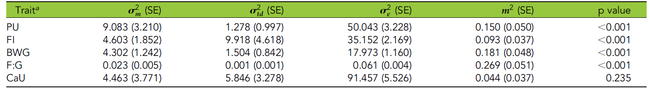
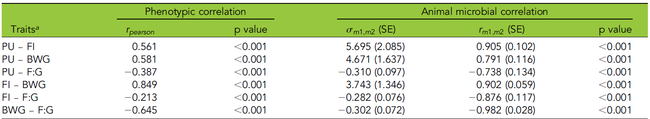
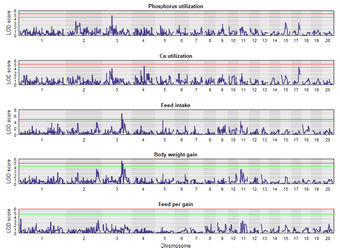
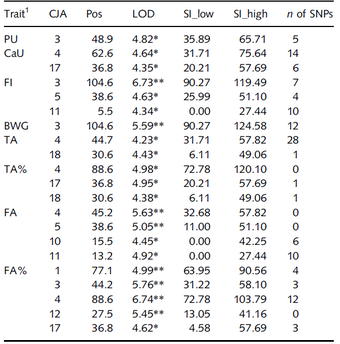
In a recent study, quantitative-genetic analyses of phosphorus (P) utilization in an F2 cross experimental design consisting of about 900 Japanese quails has been conducted. It estimated a narrow sense heritability of 14 percent. The individuals were genotyped with 4K SNP markers and lumen samples from the ileum were collected for microbiota analyses.
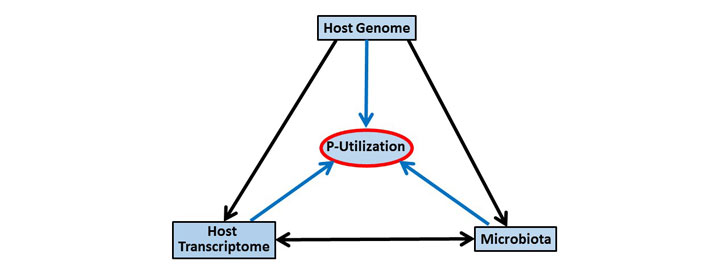
The overall aim of the present project is to obtain a better understanding of the P utilization observed in the study design considering the quail genetics, the quail ileum microbiota and the interplay between these two components. The aim is to test the following hypotheses.
- Variation of P utilization in quails is a heritable trait, which is controlled by few quail QTL with large effects.
- Variation of P utilization is partly driven by the microbial community in the ileum of the quail.
- The microbial community in the ileum is a heritable host trait that can be used for breeding individuals with improved P utilization.